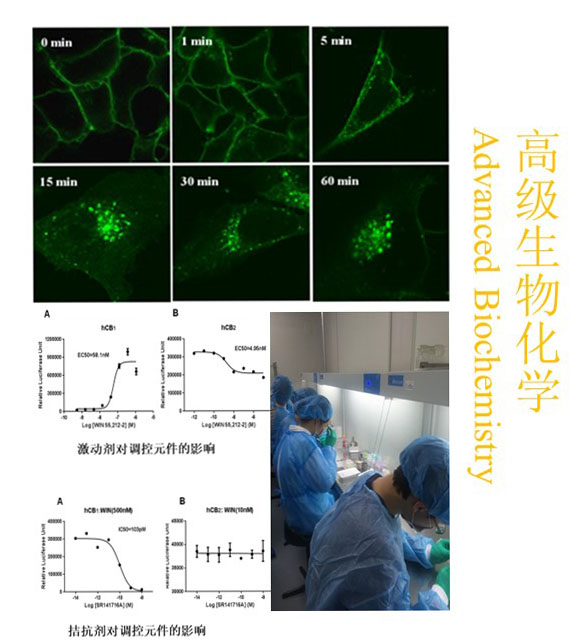
新闻与公告>>
- 中心教师参加华东五校国家级生物学实验教学... 04-23
- 2024春夏大学生物学实验教学安排 04-06
- 中心大楼成紫金港校区观花热点区 04-02
- 2024春生命科学导论实验(周日班)学生名单 03-08
- 2024春生命科学导论实验(周六班)学生名单 03-08
- 2024春生命科学导论实验(周日班)教学安排 03-08
- 2024春生命科学导论实验(周六班)教学安排 03-08
- 2024春夏生命科学导论实验(周五班)教学安排 03-05







Copyright 2003-2024 浙江大学生物学国家级实验教学示范中心 All Rrights Reserved. 技术支持
联系电话:0571-88206048 联系人:崔老师 联系邮箱:cuijin2023@zju.edu.cn
您是第 1842886 位访问者